A novel PRRT2 pathogenic variant in a family with Paroxysmal Kinesigenic Dyskinesia and Benign Familial Infantile Seizures
Abstract
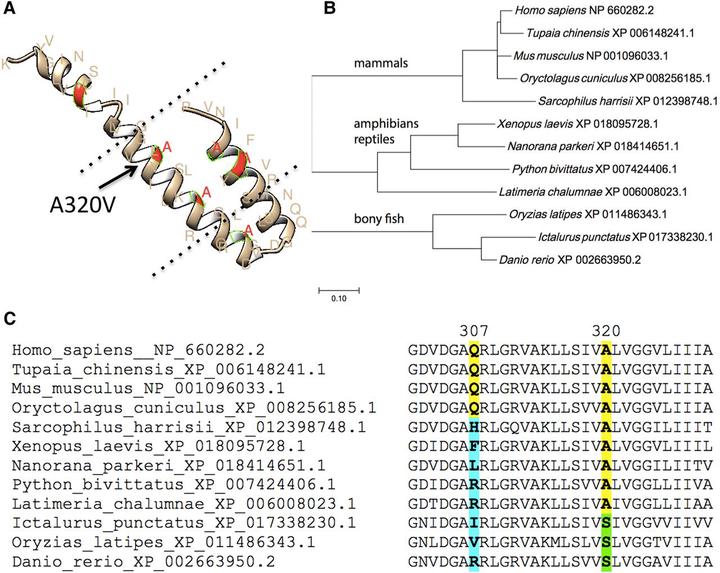
Paroxysmal Kinesigenic Dyskinesia (PKD) is a rare neurological disorder characterized by recurrent attacks of dyskinetic movements without alteration of consciousness that are often triggered by the initiation of voluntary movements. Whole exome sequencing has revealed a cluster of pathogenic variants in PRRT2 (proline-rich transmembrane protein), a gene with a function in synaptic regulation that remains poorly understood. Here, we report the discovery of a novel PRRT2 pathogenic variant inherited in an autosomal dominant pattern in a family with PKD and Benign Familial Infantile Seizures (BFIS). After targeted Sanger sequencing did not identify the presence of previously described PRRT2 pathogenic variants, we carried out whole exome sequencing in the proband and her affected paternal grandfather. This led to the discovery of a novel PRRT2 variant, NM_001256442:exon3:c.C959T/NP_660282.2:p.A320V, altering an evolutionarily conserved alanine at the amino acid position 320 located in the M2 transmembrane region. Sanger sequencing further confirmed the presence of this variant in four affected family members (paternal grandfather, father, brother, and proband) and its absence in two unaffected ones (paternal grandmother and mother). This newly found variant further reinforces the importance of PRRT2 in PKD, BFIS, and possibly other movement disorders. Future functional studies using animal models and human pluripotent stem cell models will provide new insights into the role of PRRT2 and the significance of this variant in regulating neural development and/or function.