Comparative study of the effect of disease causing and benign mutations in position Q92 on cholesterol binding by the NPC1 N-terminal domain
Abstract
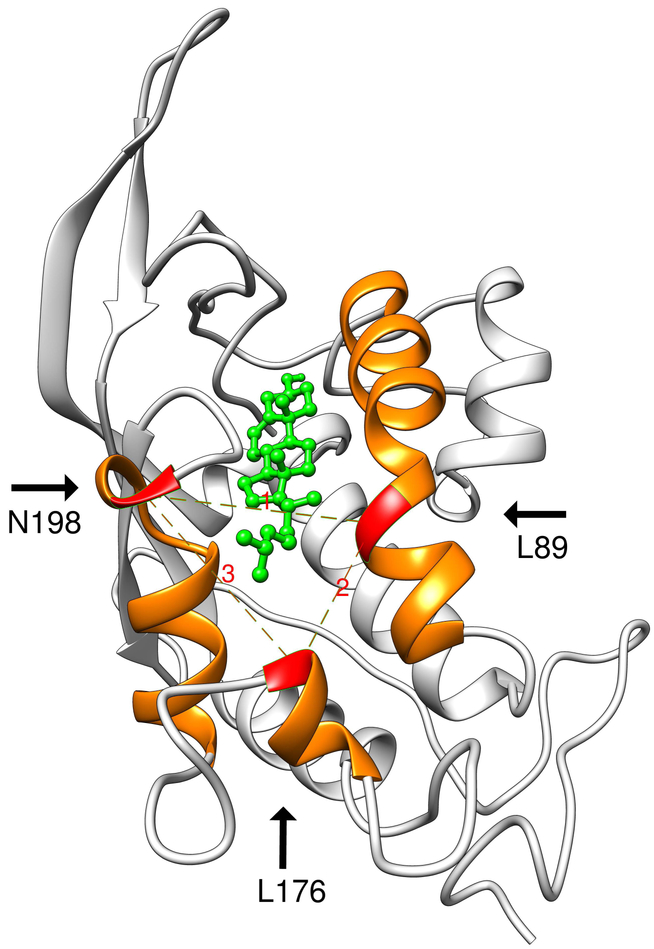
The Niemann-Pick type C1 (NPC1) protein is a large transmembrane protein located in lysosomes/endosomes. NPC1 binds cholesterol (CLR) and transports it to cellular membrane and endoplasmic reticulum. Mutations in NPC1 cause Niemann-Pick type C (NPC) disease, a rare autosomal disorder characterized by intracellular accumulations of CLR and subsequent neurodegeneration leading to premature death. Among known disease-causing mutations in NPC1, Q92R is the one that is located in the N-terminal cholesterol-binding domain [NTD]. Here we study the effect of the mutation on the ability of NPC1 (NTD) to bind and retain CLR in the binding pocket using structural analysis. We compare characteristics of the Q92R and Q92S mutant type (MT) protein, which is predicted to be benign. We provide detailed investigation of the CLR-NPC1 (NTD) binding process; and propose the mechanism, by which Q92R mutation causes NPC disease. We show that although Q92 residue neither directly participates in catalytic activity of the NPC1 (NTD), nor defines its CLR-binding specificity - it is important for the overall protein structure as well as for providing favorable electrostatic environment for CLR transfer. Our results suggest that a negative electrostatic potential of the CLR binding site (the S-opening) might promote NPC2 interaction with NPC1 (NTD) and/or proper CLR orientation and its enforced transfer. We show that in contrast to the benign Q92S mutation, Q92R significantly reduces electrostatic potential around S-opening, and thus likely affects NPC1 (NTD)-NPC2 interaction and/or CLR transfer from NPC2 to NPC1.