TREND: a platform for exploring protein function in prokaryotes based on phylogenetic, domain architecture and gene neighborhood analyses
Abstract
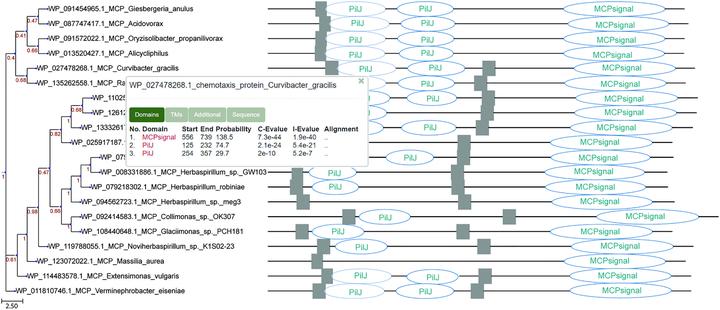
Key steps in a computational study of protein function involve analysis of (i) relationships between homologous proteins, (ii) protein domain architecture and (iii) gene neighborhoods the corresponding proteins are encoded in. Each of these steps requires a separate computational task and sets of tools. Currently in order to relate protein features and gene neighborhoods information to phylogeny, researchers need to prepare all the necessary data and combine them by hand, which is time-consuming and error-prone. Here, we present a new platform, TREND (tree-based exploration of neighborhoods and domains), which can perform all the necessary steps in automated fashion and put the derived information into phylogenomic context, thus making evolutionary based protein function analysis more efficient. A rich set of adjustable components allows a user to run the computational steps specific to his task. TREND is freely available at http://trend.evobionet.com