Research
Our lab studies how bacteria sense their environment and convert sensory inputs into behavior and physiology. We combine comparative genomics, structural inference, systems-level analysis, and evolution to predict sensory functions of signaling proteins across diverse microbes. A major emphasis is understanding the diversity and evolution of bacterial sensory receptors, including the sensor domains that recognize key metabolites, host-derived ligands, and other environmental cues.
What bacteria sense. A central goal of our research is to map sensory repertoires across microbial genomes and microbiomes. We develop frameworks that connect receptor/domain architecture to specific signals and biological function.
Evolution and diversification of sensor domains. We investigate the origins and functional specialization of widespread sensor domains, especially those of PAS and Cache superfamilies, to understand how new sensing capabilities emerge and spread across lineages. Our studies integrate evolutionary reconstruction with function-driven domain classification.
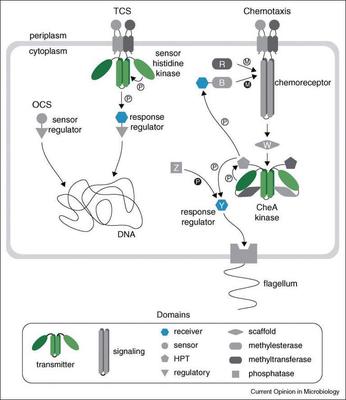
Bacterial behavior. We study chemosensory pathways and receptor families that control motility, taxis, colonization, and host interactions, including specialized sensors in important bacteria such as human pathogens and plant-associated microbes. We also investigate conserved components of the bacterial flagellar apparatus and how diversification of core proteins contributes to lineage-specific motility strategies and secretion-system function.
Major trends in bacterial signal transduction. We aim to define generalizable trends that govern how bacteria detect signals, encode information, and implement adaptive responses across diverse environments and lifestyles. We focus on the major molecular components that form the backbone of bacterial signaling networks - chemoreceptors, histidine kinases, second-messenger turnover enzymes, and transcription factors - to understand the roles these systems play across bacteria in general, as well as in specific environments and taxonomic contexts.
Community resources and foundational infrastructure. We maintain and expand community bioinformatics resources supporting microbial signal-transduction research, including database releases and platforms that enable domain- and genome context-based functional exploration.